Casgevy : le premier médicament CRISPR qui éteint la drépanocytose
Pendant presque toute l'histoire humaine, le mot « incurable » était une porte fermée. On naissait avec les gènes qu'on avait reçus, et la conversation s'arrêtait là. Puis, le 8 décembre 2023, l'agence américaine du médicament (la FDA) a discrètement entrebâillé cette porte. Elle a approuvé un médicament appelé Casgevy — le tout premier traitement bâti sur CRISPR, cet outil de « rechercher-remplacer » moléculaire qui permet aux scientifiques de réécrire le code du vivant lui-même. La maladie visée est la drépanocytose, un trouble sanguin héréditaire qui torture les humains depuis qu'il existe des humains. Et dans l'essai qui a valu l'autorisation, il n'a pas seulement aidé. Pour presque tout le monde, il a fait taire la douleur.
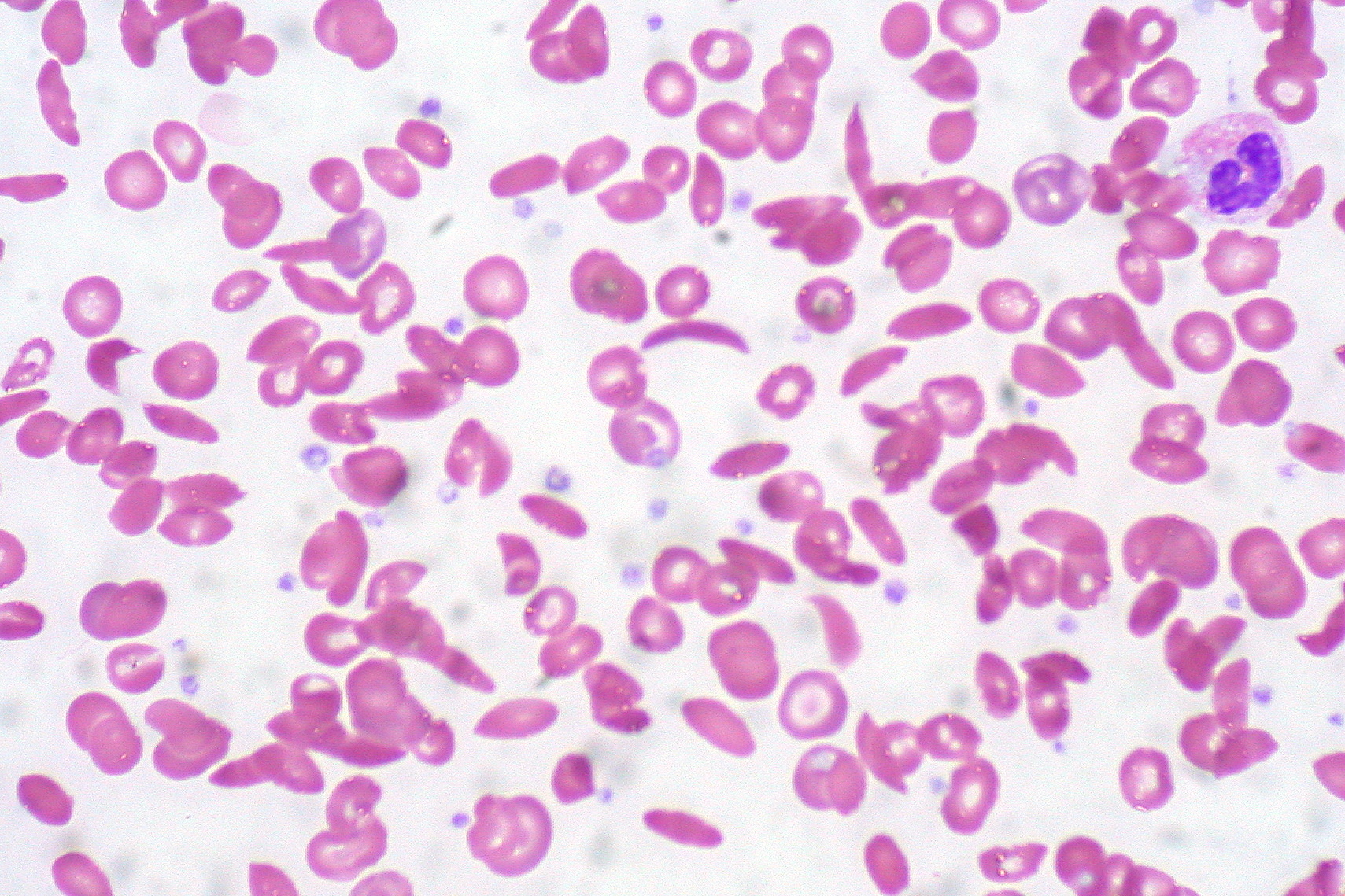
Une faute de frappe, écrite dans le sang
À sa racine, la drépanocytose est une simple faute de frappe. Une seule lettre erronée dans le gène de l'hémoglobine — la protéine qui transporte l'oxygène dans vos globules rouges — fait que cette protéine s'agglutine quand l'oxygène vient à manquer. Les globules rouges sains sont des disques souples et ronds qui se faufilent dans les plus fins vaisseaux. Les globules drépanocytaires deviennent rigides et en forme de croissant, comme de minuscules éclats de verre. Ils s'accrochent, s'empilent et bloquent la circulation. Le résultat est une « crise vaso-occlusive » : des vagues de douleur si intenses que les patients les décrivent comme des os qu'on écrase, et qui les envoient souvent à l'hôpital pendant des jours. La maladie touche environ 100 000 personnes aux États-Unis, en immense majorité noires, et a longtemps reçu une fraction de l'argent de recherche consacré à des affections plus rares.
Et voici le retournement cruel qui rend le remède si malin. Nous savons tous, en naissant, fabriquer une autre version de la protéine, parfaitement bonne : l'hémoglobine fœtale. C'est elle qui transporte l'oxygène dans le ventre maternel. Mais peu après la naissance, un interrupteur génétique l'éteint et allume l'hémoglobine adulte — et pour les patients drépanocytaires, « adulte » signifie « défectueuse ». La version fœtale, elle, n'a jamais eu la faute de frappe.
Éditer l'interrupteur
Les scientifiques se sont alors posé une question magnifique : et si on rallumait simplement l'interrupteur ?
Casgevy ne cherche pas du tout à réparer le gène de l'hémoglobine défectueux. Il utilise plutôt CRISPR-Cas9 — une paire de ciseaux moléculaires empruntée au système immunitaire des bactéries — pour faire une coupure précise dans un autre gène, BCL11A, justement celui qui sert d'interrupteur d'extinction de l'hémoglobine fœtale. Désactivez cet interrupteur dans les cellules souches sanguines du patient, et ces cellules se remettent à produire de l'hémoglobine fœtale. La bonne protéine afflue de nouveau. Les croissants ne se forment plus.
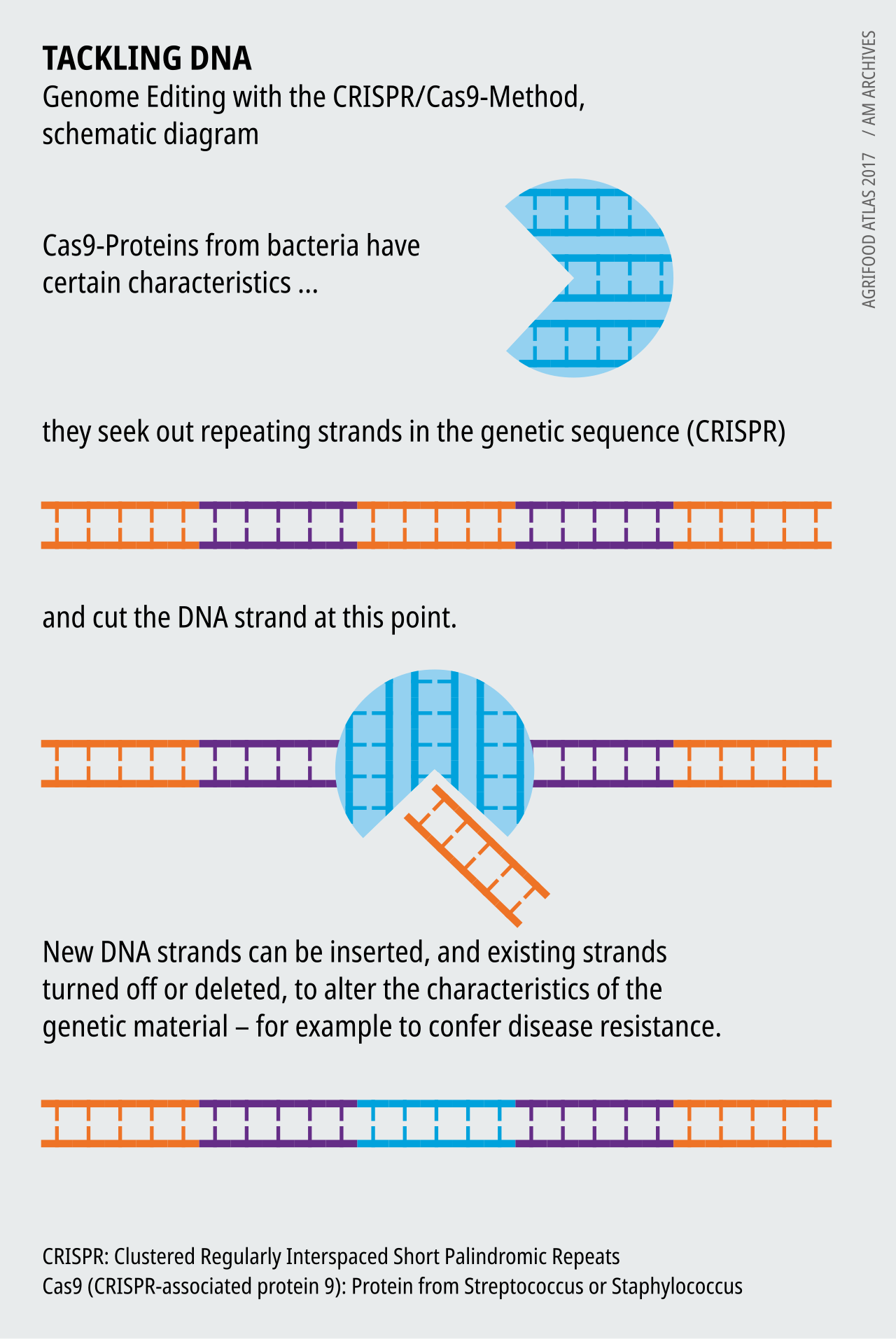
Le traitement n'est pas un cachet, et il n'a rien de doux. Les médecins prélèvent les propres cellules souches sanguines du patient, les envoient dans un laboratoire où CRISPR les réécrit, puis utilisent une chimiothérapie lourde (un médicament appelé busulfan) pour anéantir l'ancienne moelle osseuse défaillante — afin de faire de la place aux cellules éditées, qui reviennent reconstruire toute la réserve de sang de zéro. Le parcours complet prend la majeure partie d'une année, dont plusieurs semaines à l'hôpital. Mais comme l'édition vit dans les cellules souches, elle est censée durer toute une vie. Une seule fois. Terminé.
« Je peux rêver à nouveau »
Les chiffres de l'essai décisif sont de ceux qui font sursauter les hématologues. Sur les 30 patients suivis assez longtemps pour être évalués, 29 — soit 97 % — ont passé au moins 12 mois d'affilée sans la moindre crise vaso-occlusive. Les 30 sont tous restés hors de l'hôpital, sans hospitalisation pour crise, pendant au moins un an. Pour des gens dont la vie était organisée autour de la prochaine vague de douleur inévitable, ce n'est pas une amélioration. C'est une autre vie.
La pionnière fut une femme nommée Victoria Gray, du Mississippi, qui a reçu la version expérimentale dès juillet 2019. Elle raconte qu'il lui a fallu environ huit mois pour vraiment prendre la mesure du changement — un matin, elle s'est réveillée sans douleur et a cru que son corps était simplement engourdi. « À un moment de ma vie, j'avais cessé de faire des projets, parce que je sentais que je n'avais pas d'avenir », a-t-elle confié lors d'un sommet sur l'édition génétique. « Aujourd'hui, je peux rêver à nouveau, sans limites. »

Le hic, et l'horizon
Il y a, bien sûr, un hic — et il est de taille. Le prix affiché de Casgevy tourne autour de 2,2 millions de dollars, et l'ensemble du parcours de soins, avec la chimiothérapie et les mois de suivi, peut rapprocher le coût réel des 3 millions. Un vrai remède existe pour une maladie qui frappe, en grande majorité, ceux qui sont le moins en mesure de le payer. Cette tension — entre ce que la science sait désormais faire et ceux qui y ont réellement accès — est l'énigme qui définit l'ère de l'édition génétique.
Mais prenez du recul et regardez ce qui vient de se produire. Un mécanisme de défense bactérien, détourné par l'humain il y a à peine une décennie, a servi à réécrire un seul point ciblé du génome d'une personne et à lever une condamnation à la douleur à vie. La même approche vise déjà d'autres maladies héréditaires, de la cécité au cholestérol élevé. Casgevy n'est pas la fin de l'histoire. C'est la preuve que la porte, jadis fermée pour toujours, s'ouvre vraiment.